 |
|
|
|
!! Tips !!
Wear a glove and avoid RNase activity in general. All the manipulations should be done on ice.
1. Dissolve 100 ug of the RNA in 100 uL of dH2O.
2. Incubate at 60°C for 3 min to denature.
3. Set up a BAP reaction by combining:
| sample | 100uL |
| dH2O | 39.6 uL |
| 5 x BAP buffer | 5.4 uL |
| RNasin | 2.0 uL |
| BAP (0.4U/uL) | 12.5 uL |
4. Incubate at 37°C for 60 min.
5. Add an equal volume of phenol : chloroform (1 : 1) to the sample and mix well. Centrifuge at 14,000 rpm briefly at 4°C. Transfer the upper aqueous layer to a fresh tube.
6. Ethanol precipitate the RNA by adding:
| ethachinmate | 1 uL |
| sodium acetate, pH 5.5 | 20 uL |
| 100% ethanol | 500 uL |
Centrifuge at 14,000 rpm at 4°C for 10 min.
7. Remove the supernatant and rinse the pellet with 100 uL of 80% (v/v) ethanol. Drying is not necessary.
8. Dissolve the sample in 36.65 uL of dH2O.
|
|
|
|
|
| STEP03. TAP Reaction |
Protocol |
|
|
|
|
|
|
!! Tips !!
Wear a glove and avoid RNase activity in general. All the manipulations should be done on ice.
1. Set up a TAP reaction by combining:
| sample | 36.65 uL |
| 5 x TAP buffer | 10.0 uL |
| RNasin | 1.35 uL |
| TAP | 2.0 uL |
2. Incubate at 37°C for 60 min.
3. Add 50 uL of dH2O.
4. Add an equal volume of phenol : chloroform (1 : 1) to the sample and mix well. Centrifuge at 14,000 rpm briefly at 4°C. Transfer the upper aqueous layer to a fresh tube.
5. Ethanol precipitate the RNA by adding:
| ethachinmate | 1 uL |
| sodium acetate, pH 5.5 | 10 uL |
| 100% ethanol | 250 uL |
6. Centrifuge at 14,000 rpm at 4°C for 10 min. Remove the supernatant and rinse the pellet with 100 uL of 80% (v/v) ethanol. Drying is not necessary.
7. Re-suspend the sample in 21.0 uL of dH2O.
|
|
|
|
|
|
|
| STEP04. RNA Ligation |
Protocol |
|
|
|
|
|
|
!! Tips !!
Wear a glove and avoid RNase activity in general. All the manipulations should be done on ice.
1. Ligate the BAP/TAP-treated poly(A)+ RNA to the 5'-oligoribonucleotide by combining:
| sample | 21.7 uL |
| 5'-oligoribonucleotide (100 ng/uL) | 12.0 uL |
| 10 x ligation buffer | 30.0 uL |
| 50 mM MgCl2 | 60.0 uL |
| 24 mM ATP | 6.3 uL |
| RNasin | 7.5 uL |
| T4 RNA ligase (40U/uL) | 12.5 uL |
| 50 % (w/v) PEG 8000 | 150.0 uL |
2. Incubate at 20°C for 3 h.
3. Add 450 uL of dH2O.
4. Extract with 300 uL of phenol : chloroform (1 : 1). Ethanol precipitate by adding 1 uL of ethachinmate, 60 uL of sodium acetate, pH 5.5 and 1500 uL of 100% ethanol (the rest of the procedure is as described in 3.6).
5. Dissolve the sample in 54.3 uL of dH2O.
|
|
|
|
|
|
|
| STEP05. DNase I Treatment |
Protocol |
|
|
|
|
|
|
!! Tips !!
DNaseI treatment can be omitted if contaminating genomic DNA is negligible.
1. Remove the residual DNA with DNase I by combining:
| sample | 54.3 uL |
| 25 mM MgCl2 | 32.0 uL |
| 1M Tris-HCl (pH 7.0) | 4.0 uL |
| 0.1 M dithiothreitol (DTT) | 5.0 uL |
| RNasin | 2.7 uL |
| DNase I | 2.0 uL |
*Use DTT supplied with SuperScript II
2. Incubate at 37°C for 10 min.
3. Extract with phenol : chloroform (1 : 1) and ethanol precipitate (as described in 3.4-6).
4. Dissolve the sample in 1200 uL of dH2O.
|
|
|
|
|
|
|
| STEP06. Poly(A) Selection of the RNA with Oligo-dT cellulose |
Protocol |
|
|
|
|
|
|
1. Transfer the oligo-dT powder from two pre-packed columns* to a polypropylene Poly-Prep column (BIORAD). The bed volume of the powder should be approximately 0.5 mL when the powder from two columns is used.
2. Denature the dT powder by washing with 3 mL of 0.1 N NaOH.
3. Wash out the alkaline solution with 5 mL of dH2O.
4. Pre-equilibrate the column with 5 mL of 1 x Loading Buffer.
5. Set a fresh tube to collect the flow through.
6. Add an equal volume (1.2 mL) of 2x Loading Buffer to the sample, mix well and apply to the column.
7. Collect the flow through and apply to the column. Repeat this step two more times.
8. Wash the column with 5 mL of 1x Loading Buffer.
9. Set a fresh collection tube and elute the sample by applying 3 mL of dH2O.
10. Add 8 mL of 100% ethanol and 330 uL of 3 M sodium acetate (pH 5.5) and centrifuge for ethanol precipitation.
11. Dissolve the sample in 100 uL of dH2O and ethanol precipitate once more (as described in 3.5-6).
12. Dissolve the sample in 21.0 uL of dH2O.
|
|
|
|
|
|
|
| STEP07. First-Strand cDNA Synthesis |
Protocol |
|
|
|
|
|
|
1. Transfer the oligo-dT powder from two pre-packed columns* to a polypropylene Poly-Prep column (BIORAD). The bed volume of the powder should be approximately 0.5 mL when the powder from two columns is used.
| sample | 21.0 uL |
| 5 x First strand buffer | 10.0 uL |
| 4 dNTPs at 5 mM each | 8.0 uL |
| 0.1M DTT | 6.0 uL |
| oligo dT adapter primer | 2.5 uL |
| RNasin | 1.0 uL |
| SuperScript II | 2.0 uL |
for the TSS/PAS Mate Pair library. For the TSS/Random Mate Pair library, use 2.5 uL of the dR (random hexamer) adapter primer instead of the oligo dT adapter primer.
* It may be better to utilize a different oligonucleotide that harbours a V (mix of C,G,A) at the 3' terminal position. The oligonucleotide would exclude amplification initiated within long 3' terminal poly(A) stretches, although we have not tried this option.
2. Incubate at 42°C for more than 3 h for the TSS/PAS Mate Pair library. For the TSS/Random Mate Pair library, incubate at 12°C for 1 h and 42°C for more than 3 h.
* Note: Random hexamer oligonucleotide terminates in cytosine at 3' ends, as we intended to enhance the hybridization at the starting site of the reverse transcription because the binding of G-C pair is stronger than that of A-T pair.
3. Add 50 uL of dH2O and extract the solution with phenol : chloroform (1 : 1) (as described in 3.4).
4. Add 2 uL of 0.5 M EDTA pH 8.0, to thoroughly stop the reaction.
|
|
|
|
|
|
|
| STEP08. Alkaline Degradation of the Template mRNA |
Protocol |
|
|
|
|
|
|
1. Degrade the template RNA by adding 15 uL of 0.1 M NaOH. Incubate at 65°C for 40 min.
2. Add 20 uL of 1 M Tris-HCl, pH 7.0 to neutralize.
3. To remove the fragmented RNA, ethanol precipitate the first strand cDNA by adding:
| ethachinmate | 1 uL |
| 7.5 M ammonium acetate | 70 uL |
| ethanol | 500 uL |
(the rest of the procedure is as described in 3.6).
4. Dissolve the sample in 31 uL of dH2O.
5. Check 1 uL of the DNA with a bioanalyzer.
|
|
|
|
|
|
|
| STEP09. PCR Amplification of the cDNA |
Protocol |
|
|
|
|
|
|
1. Use 10 uL for TSS/PAS Mate Pair library, 20 uL for TSS/Random Mate Pair library
| sample + dH2O | 52.4 uL |
| 3.3 x Reaction buffer II | 30.0 uL |
| 25 mM magnesium acetate | 4.4 uL |
| PCR 5'-primer | 1.6 uL |
| PCR 3'-primer | 1.6 uL |
| DNA polymerase | 2.0 uL |
(the rest of the procedure is as described in 3.6).
2. Thermocycle for 20 cycles at 94°C, 1 min; 58°C, 1 min; 72°C, 2 min.
3. Extract with phenol : chloroform (1 : 1) and ethanol precipitate (as described in 2.3.4-6).
4. Dissolve the sample in 10 uL of dH2O.
!! Tips !!
If the size of the amplicon is less than 1kb on average, efficacy of the cap replacement may have been failed.
Bioanalyzer chart of PCR product
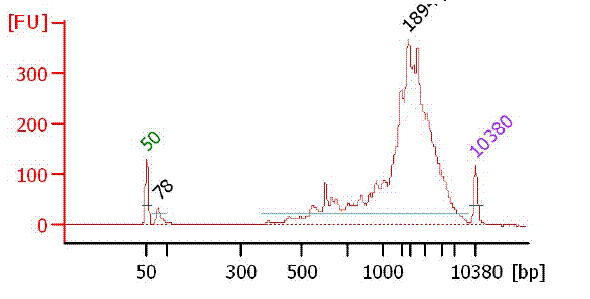
|
|
|
|
|
|
|
| STEP10. Size Fractionation of the PCR products |
Protocol |
|
|
|
|
|
|
1. Prepare a 1% agarose gel
2. Run gel at 100 V for 30 min.
3. Excise a chunk of gel containing the DNA fraction from 500 bp to 5 kbp for the TSS/PAS Mate Pair library. Excise three chunks of gel containing the DNA fractions from 500 bp to 1 kbp, from 1 kbp to 2 kbp, and from 2 kbp to 5 kbp for the TSS/Random Mate Pair library.
4. Purify the DNA with Gel Extraction Kit.
5. Check the DNA with a bioanalyzer.
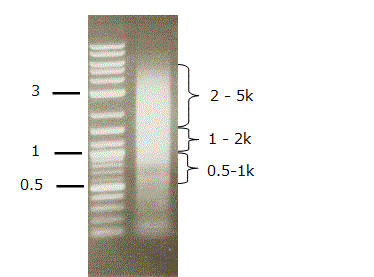
Picture of TSS/PAS PCR product on 1% agarose gel

Bioanalyzer chart of TSS/PAS gel fraction (0.5 - 5kbp)
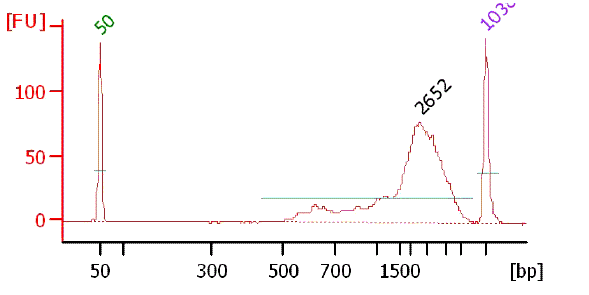
Picture of TSS/Random PCR product on 1% agarose gel
Bioanalyzer chart of TSS/Random gel fraction (500bp - 1k)
Bioanalyzer chart of TSS/Random gel fraction (1k - 2k)
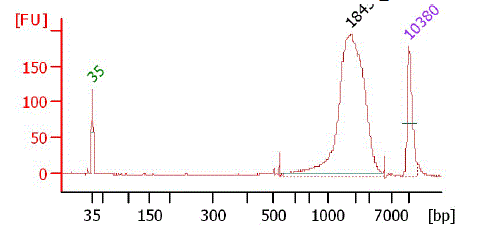
Bioanalyzer chart of TSS/Random gel fraction (2k - 5k)
|
|
|
|
|
|
|
| STEP11. Perform End Repair |
Protocol |
|
|
|
|
|
|
1. Dissolve 600 ug of the DNA in 75 uL of dH2O.
| Size selected DNA | 75 uL |
| 10 x End Repair Buffer | 10 uL |
| Water | 7.5 uL |
| Natural dNTP Mix | 4 uL |
| T4 DNA Polymerase | 5 uL |
| T4 Polynucleotide Kinase | 5 uL |
| Klenow DNA Polymerase | 1 uL |
(the rest of the procedure is as described in 3.6).
2. Incubate at 20°C for 30 min. After the incubation place immediately on ice.
3. Purify the DNA with QIAquick PCR Purification Kit. Dissolve the sample in 50 uL of dH2O.
|
|
|
|
|
|
|
| STEP12. Circularize DNA |
Protocol |
|
|
|
|
|
|
1. Circularize the DNA to bind the TSS and PAS for TSS/PAS Mate Pair library, or to bind the TSS and inside cDNA for TSS/Random Mate Pair Library.
| Sample | 50 uL |
| 10 x circularization buffer | 30 uL |
| dH2O | 206.6 uL |
| Circularization ligase | 13.4 uL |
(the rest of the procedure is as described in 3.6).
2. Incubate overnight for 16 h at 30°C.
|
|
|
|
|
|
|
| STEP13. Digest Linear DNA (Remove the linear DNA with DNA Exonuclease.) |
Protocol |
|
|
|
|
|
|
1. Add 3 uL of DNA exonuclease to 300 uL of the circularization reaction. Mix by gently flicking the tube and briefly centrifuge.
2. Incubate at 37°C for 20 minutes followed by 70°C for 30 minutes.
3. Add 12 uL of EDTA to the exonuclease-treated sample.
4. Add an equal volume of phenol : chloroform (1 : 1) to the sample and mix well. Centrifuge at 14,000 rpm briefly at 4°C. Transfer the upper aqueous layer to a fresh tube.
5. Ethanol precipitate the RNA by adding:
| ethachinmate | 1 uL |
| 3M sodium acetate | 30 uL |
| 100 % ethanol | 750 uL |
6. Dissolve the sample in 37.5 uL of dH2O.
|
|
|
|
|
|
|
| STEP14. Fragment Circularized DNA |
Protocol |
|
|
|
|
|
|
1. Fragment the circularized DNA at the EcoP 15I recognition site.
| Sample | 37.5 uL |
| 10 x NEBuffer | 5 uL |
| 100 x BSA | 0.5 uL |
| 10 x ATP | 5 uL |
| EcoP15I | 2 uL |
1-1. Incubate at 37°C for 1 h.
1-2. The EcoP enzyme was inactivated at 65°C for 20 min.
1-3. Add 50 uL of dH2O.
1-4. Extract with phenol : chloroform (1 : 1) and ethanol precipitate (as described in 3.4-6).
1-5. Dissolve the sample in 50 uL of dH2O.
2. For the TSS/PAS Mate Pair library, additionally fragment with nebulization.
2-1. Transfer EcoP15I-treated DNA to the nebulizer, and add 250 uL of dH2O, 400 uL of the nebulization buffer and approximately 150 uL of 100% glycerol to the DNA and mix well.
2-2. Chill the nebulizer containing the DNA solution on ice while performing the next step.
2-3. Nebulize with delivered air at 32 psi for 6 min. Vapor may rise from the nebulizer, which is normal.
2-4. Centrifuge the nebulizer at 450 xg for 2 minto collect the approximately 400 uL of droplets from the side of the nebulizer.
2-5. Follow the instructions in the QIAquick PCR Purification Kit to purify the sample solution and concentrate it on one QIAquick column, elutingin 50 ul of dH2O.
!! Tips !!
Proper size range of the fragmented DNA should be confirmed.
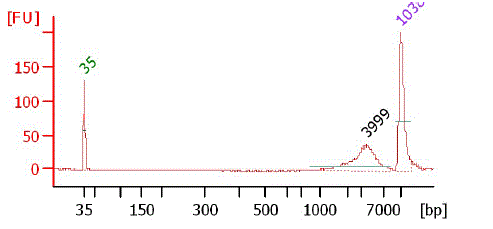
Bioanalyzer chart after nebulization
|
|
|
|
|
|
|
| STEP15. Purify Biotinylated DNA |
Protocol |
|
|
|
|
|
|
Select the fragmented DNA with biotin using the Dynal magnetic M-280 streptavidin beads.
1. Transfer 20 uL of well resuspended beads into a 1.5-mL microcentrifuge tube.
2. Place the tube in the magnetic rack for about 1 min, until the beadsare separated from the solution. Remove and discard the supernatant.
3. Wash the beads. Add the 50 ul of streptavidin bead binding buffer. Add the buffer solution and resuspend the beads by gently flicking the tube. Centrifuge the tube for 1-2 s. Place the tube in the magnetic rack for 1 min, then remove and discard the supernatant.
4. Repeat step 3 once.
5. Resuspend the beads in 50 uL of fresh streptavidin bead binding buffer.
6. Add 50 uL of the bead solution to the 50 ul of fragmented DNA sample and incubate for 15 min at 20°C. Resuspend the beads every 2 min by gentle mixing.
7. Place the tube in the magnetic rack for 1 min, and then remove and discard the supernatant.
8. Wash the beads in 200 uL of streptavidin bead wash buffer 1. Add the buffer solution and resuspend the beads by gently flicking the tube. Centrifuge the tube for 1-2 s. Place the tube in the magnetic rack for 1 min, then remove and discard the supernatant.
9. Repeat step 8 three times.
10. Wash the beads in 200 uL of QIAGEN EB Buffer. Add the buffer solution and resuspend the beads by gently flicking the tube. Centrifuge the tube for 1-2 s. Place the tube in the magnetic rack for 1 min, then remove and discard the supernatant.
11. Wash the beads for a second time in 200 uL of QIAGEN EB buffer. Place the sample on ice without removing the final wash solution.
|
|
|
|
|
|
|
| STEP16. Perform End Repair |
Protocol |
|
|
|
|
|
|
Convert the overhangs resulting from fragmentation into blunt ends.
1. Prepare the reaction mix in a new tube on ice.
| 10 x End Repair Buffer | 10 uL |
| dH2O | 75 uL |
| Natural dNTP Mix | 4 uL |
| T4 DNA Polymerase | 5 uL |
| T4 Polynucleotide Kinase | 5 uL |
| Klenow DNA Polymerase | 1 uL |
2. Place the washed beads back on the magnet for 1 min; remove and discard the supernatant.
3. Resuspend the beads immediately in 100 uL of the end-repair reaction mix.
4. Incubate for 30 mins at 20°C.
5. Place the tube in the magnetic rack for 1 min; remove and discard the supernatant.
6. Wash the beads in 200 uL of streptavidin bead wash buffer 1 as described in 15.8. and 15.9.
7. Wash the beads in 200 uL of QIAGEN EB buffer as described in 15.10.
8. Wash the beads for a second time in 200 uL of QIAGEN EB buffer. Place the sample on ice without removing the final wash solution.
|
|
|
|
|
|
|
| STEP17. A-tail DNA Fragment |
Protocol |
|
|
|
|
|
|
Add an 'A' base to the 3'-ends of the blunt phosphorylated DNA fragments.
1. Prepare the reaction mix in a new tube.
| 10qۢ-Tailing buffer | 5 uL |
| dH2O | 32 uL |
| 1mM dATP | 10 uL |
| A-Tailing enzyme | 3 uL |
2. Place the washed beads back on the magnet for 1 min; remove and discard the supernatant.
3. Resuspend the beads immediately in 50 uL of A-Tailing reaction mix.
4. Incubate for 30 min at 37°C.
5. Place the tube in the magnetic rack for 1 min; remove and discard the supernatant.
6. Wash the beads in 200 uL of streptavidin bead wash buffer 1 as described in 15.8. and 15.9.
7. Wash the beads in 200 uL of QIAGEN EB buffer as described in 15.10.
8. Wash the beads for a second time in 200 uL of QIAGEN EB buffer. Place the sample on ice without removing the final wash solution.
|
|
|
|
|
|
|
| STEP18. Ligate Adapters to DNA Fragments |
Protocol |
|
|
|
|
|
|
1. Prepare the reaction mix in a new tube.
| 2 x Adapterligation Buffer | 25 uL |
| dH2O | 19 uL |
| PE Adapter oligo Mix | 1 uL |
2. Place the washed beads back on the magnet for 1 min; remove and discard the supernatant.
3. Resuspend the beads immediately in 45 uL of A-Tailing reaction mix.
4. Add 5 uL of adapter ligase to the reaction and mix by pipetting gently up and down several times.
5. Incubate for 15 min at 20°C.
6. Wash the beads in 200 uL of streptavidin bead wash buffer 1 as described in 15.8. and 15.9.
7. Wash the beads in 200 uL of streptavidin bead wash buffer 2. Add the buffer solution and resuspend the beads by gently flicking the tube. Centrifuge the tube for 1-2 s. Place the tube in the magnetic rack for 1 min, then remove and discard the supernatant. Repeat wash once, discarding wash supernatant.
8. Wash the beads in 200 uL of QIAGEN EB buffer as described in 15.10.
9. Wash the beads for a second time in 200 uL of QIAGEN EB buffer. Place the sample on ice without removing the final wash solution.
|
|
|
|
|
|
|
| STEP19. Enrich Adapter-Modified DNA Fragments by PCR |
Protocol |
|
|
|
|
|
|
1. Prepare the following PCR mix.
| Phusion DNA polymerase | 25 uL |
| dH2O | 23 uL |
| PCR Primer 1.0 | 1 uL |
| PCR Primer 2.0 | 1 uL |
2. Place the washed beads back on the magnet for 1 min; remove and discard the supernatant.
3. Resuspend the beads in 50 uL of the PCR mix and transfer to a 0.2 mL- PCR tube.
4. Thermocycle for 18 cycles at 98°C for 10 s; 65°C for 10 s; 72°C for 30 s.
5. Remove and retain the PCR supernatant from the beads using a magnetic rack, and discard the beads
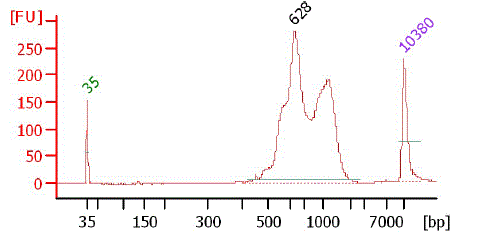
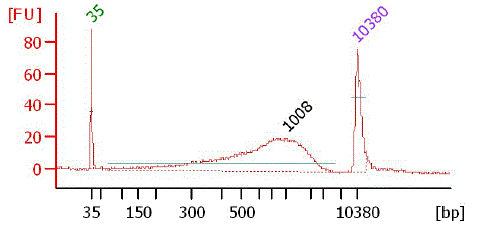
Bioanalyzer chart of PCR product
|
|
|
|
|
|
|
| STEP20. Size selection |
Protocol |
|
|
|
|
|
|
Size Fractionation of the Products
1. Prepare an 8% polyacrylamide gel.
2. Run gel at 160 V for 90 min.
3. Excise a chunk of gel containing the DNA fraction from 280 bp to 330 bp for TSS/PAS Mate Pair library, and a single band around 250 bp for TSS/Random Mate Pair library.
4. Purify the DNA and dissolve in 5 uL of dH2O.
5. Check 1uL of the DNA with a bioanalyzer.
!! Tips !!
Typical examples of the prepared templates are shown.
Picture of TSS/PAS PCR product on 8% polyacrylamide gel

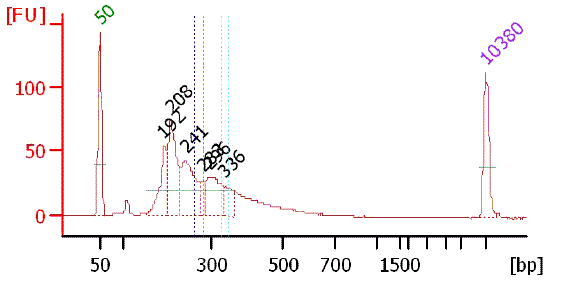
Bioanalyzer chart of purified TSS/PAS Mate Pair library
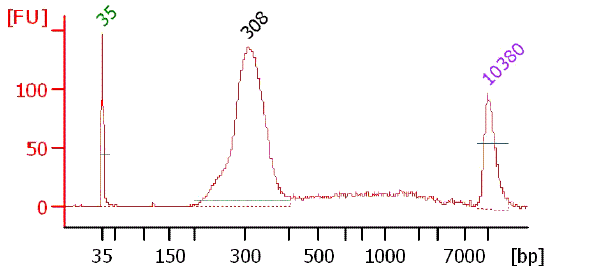
Picture of TSS/Random PCR product on 8% polyacrylamide gel

Bioanalyzer chart of purified TSS/Random Mate Pair library (500bp-1k)
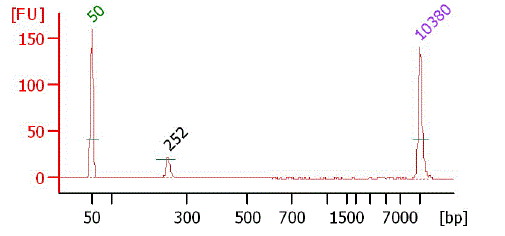
Bioanalyzer chart of purified TSS/Random Mate Pair library (500bp-1k)
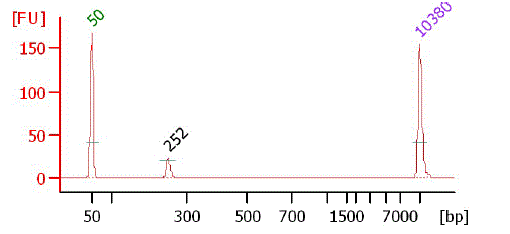
|
|
|
|
|
|
|
| STEP21. Sequencing and Mapping |
Protocol |
|
|
|
|
|
|
1. Sequencing was conducted on the Illumina HiSeq2000 platform following the manufacturer's instructions. Paired-end reads of 101 bases were generated for at least 10 million tags for each library.
2. The generated sequence tags were mapped onto the human genomic sequence (hg19 as of UCSC Genome Browser) using the sequence alignment Programme ELAND v2 (Illumina).
3. For the mapping, proper directions of the mate-pair tags should be checked (TSS and PAS tags should be mapped in a designated orientation R < F).
|
|
|
|
|
|
|
|